




For most of her life, 19-year-old Jordan Stinchcomb’s parents thought she had cerebral palsy.
Jordan was born by emergency Cesarean section with the umbilical cord wrapped around her neck, and had to be resuscitated. Her parents assumed that lack of blood flow and oxygen at birth had damaged her brain, causing her inability to walk and speak.
Pam and Tony Stinchcomb, who live in Hoschton, Georgia, had two more children, Jenna and Jake, who were born without similar difficulties. Then when Jordan was 13, her youngest sister Jessie was born, after an almost perfect pregnancy.
“We knew something was not right,” says Pam, a physical therapist. “She cried for the first six months and would scream if we put her on her stomach.”
Jessie missed early milestones such as being able to roll over or sit up by herself. At around six months of age, she started wheezing because of an airway restriction. Her parents thought they saw hints that Jessie was experiencing seizures, which Jordan had started having at age 11. They decided to seek help and an explanation.
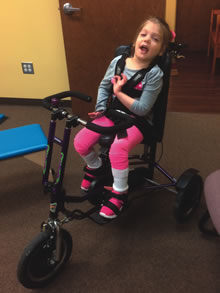
Jessie in her adaptive tricycle
Neurologists they consulted suspected Jessie had a congenital condition: perhaps Rett syndrome, a neurodevelopmental disorder that mainly affects girls, or a mitochondrial disorder. A muscle biopsy and other invasive procedures were performed. Months went by, while tests for known diseases came back negative.
When the Stinchcombs were referred to Emory, they encountered a delay when a respected geneticist who specialized in rare disorders, Paul Fernhoff, associate professor of human genetics and pediatrics and the medical director of human genetics at Emory University Hospital, died unexpectedly.
When medical geneticist Michael Gambello arrived at Emory from Houston in 2012, he recommended that the family proceed with whole exome sequencing for Jessie. It wasn’t a difficult decision. At that time, whole exome sequencing was emerging as a powerful technique for finding the mutations that might be responsible for a disease that looks like an inherited condition, when tests for known genetic disorders have not proven helpful.
Several weeks later, the Emory Genetics Laboratory, under the direction of Madhuri Hegde, found something, but it needed additional confirmation. Gambello asked whether Jordan’s DNA could also be tested.
Pam and Tony were surprised, since they had thought Jordan’s condition was caused by the circumstances of her birth.
“It made sense 19 years ago,” Pam says. “Until Jessie was born, we never had a need to look for anything else.”
Gambello told the Stinchcombs about the sequencing results, but he was able to tell them little about the disease or Jessie’s prognosis.
Pam Stinchcomb recalls the conversation going something like this:
“We’ve learned that your daughter has N-glycanase-1 deficiency. It’s only been identified recently.”
“What is that?”
“We really don’t know. There is another patient reported in the medical literature, who has symptoms almost identical to Jessie.”
The disorder was not something doctors were familiar with, even geneticists who deal with rare diseases.
“At that point, there was one publication out there describing a single patient with NGLY1 deficiency,” Gambello says. “It could have been wrong. But we had two affected siblings, and the parents were carriers.”
N-glycanase 1 is an enzyme—part of a complex apparatus that processes proteins— that cell biologists have studied for years in the laboratory. However, it was highly unusual for doctors to see not one, but two living patients in the same family who appeared to lack this enzyme.
Coincidentally, shortly after talking with the Stinchcombs, Gambello received an email from a genetic counselor he had worked with in Houston, seeking advice for another family with a child with NGLY1 deficiency.
From this communication, he and the Stinchcombs learned about several other children around the world with the same genetic deficiency, whose cases had not yet been published in medical journals.
Several of the affected families share the same exact genetic mutation in the NGLY1 genes, even though the families do not appear to be related, beyond sharing European ancestry. This may indicate a “founder effect” reaching back many generations.
Having felt isolated and alone, the families were hungry for information from the medical community and from each other.
It’s hard enough for your child to have a chronic or debilitating condition, let alone one that doctors are stumped by.
One of the fathers, Matt Might, a computer scientist at the University of Utah, wanted to find other families dealing with the same challenges.
After an odyssey of diagnostic missteps led to the identification of NGLY1 deficiency in his son, Bertrand, Might wrote the essay “Hunting down my son’s killer” on his personal website.
“Aside from severe jaundice, Bertrand was normal at birth,” Might writes. “For two months, he developed normally. At three months, his development slowed, but it was ‘within normal variations.’ By six months, he had little to no motor control. He seemed, as we described it, ‘jiggly.’ Something was wrong.”
With his post amplified by social media sites like Reddit, over the next year the Mights found nine other families with a child with NGLY1 deficiency.