A study led by Emory University School of Medicine researchers, in collaboration with researchers at Albert Einstein College of Medicine in New York and Heidelberg University in Germany, demonstrated for the first time the presence of a KIF5A mutant protein in patients and shed new light on the pathogenesis leading to Amyotrophic Lateral Sclerosis (ALS).
By confirming the expression of KIF5A mutant proteins in motor neurons derived from KIF5A ALS patients and deciphering the properties of mutant motor, the study provides a window into understanding a possible trigger of ALS. According to the authors, this could contribute to the development of new therapeutic approaches to the disease. The study is now online in the journal EMBO Reports.
ALS is a fatal progressive neurodegenerative disease characterized by loss of upper and lower motor neurons leading to paralysis and death. It is also known as Lou Gehrig’s disease, named after the famous Yankee baseball player who got the illness and had to retire in 1939 because of it.
Although considered rare, ALS is the most common motor neuron disease in adults and is growing worldwide. Most ALS cases, about 90%, are not connected to a family history of the disease and are known as sporadic. About 5 to 10% of cases are familial, meaning they are connected to a family history of ALS.
In 2018, mutations in the human kinesin family protein 5A (KIF5A) were reported to be a genetic cause of ALS in European and American cohorts of patients and were later confirmed in Asian ALS cohorts.
KIF5A is an important motor protein that transports various components (proteins, lipids and RNA, referred to as “cargo”) that are critical for neuronal health and function. The KIF5A ALS variants cause a skipping of exon 27 at the RNA level. The KIF5A mutations are predicted to produce a new protein with a neopeptide, but this has not been demonstrated in patients until this study.
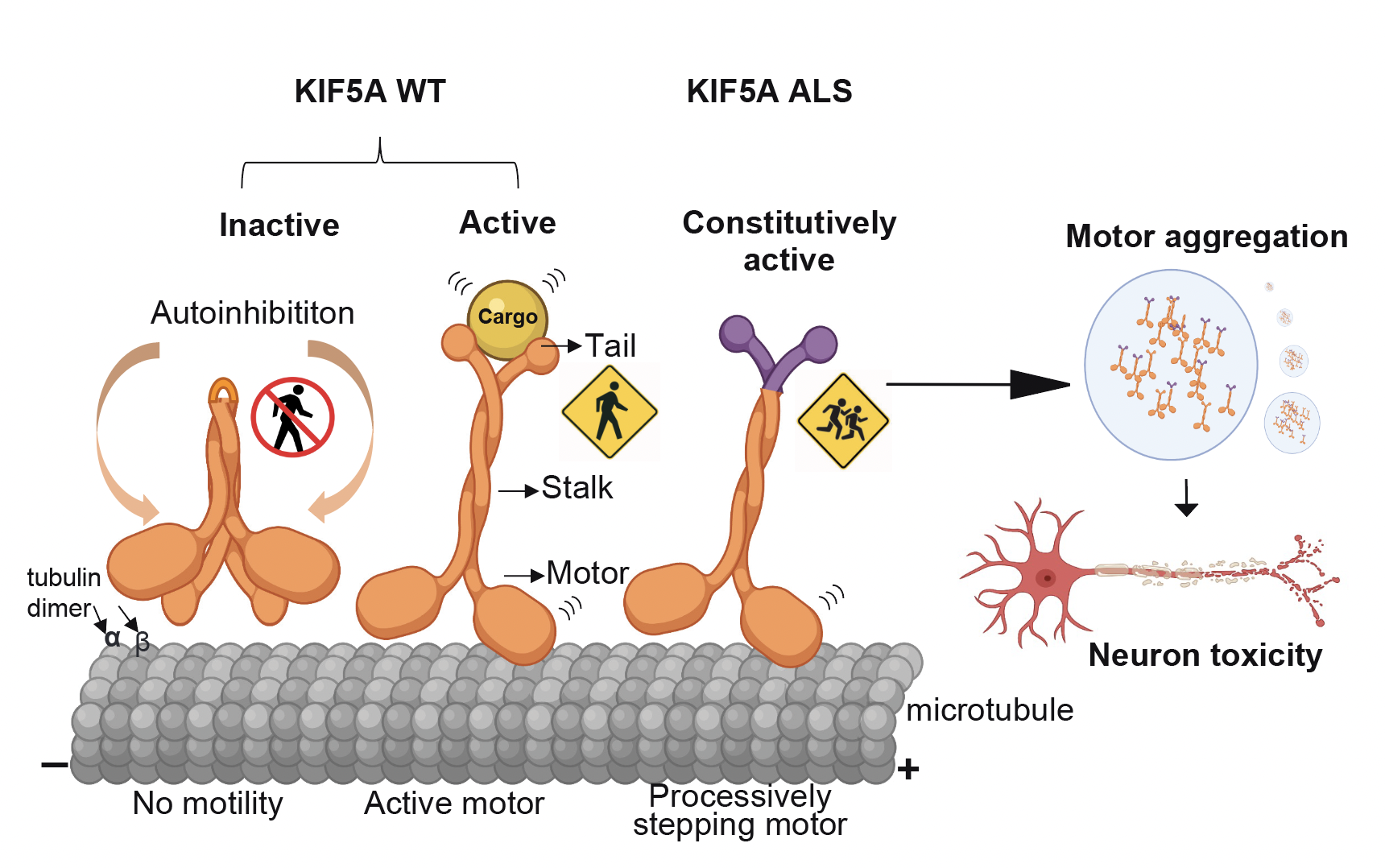
The KIF5A motor transports cargos including proteins, lipids and RNA, which are critical components for neuronal health and function. The new study reveals that ALS-associated KIF5A mutations cause neuronal toxicity via gain-of-function caused by constitutive activation and increased motor association and aggregation.
Researchers also performed a comprehensive analysis of the mutant KIF5A protein at single-molecule, cellular and organism levels. They found that mutant protein is hyperactive, forms cytoplasmic aggregates and causes toxicity in both in-vitro and in-vivo models.
The study was led by Devesh Pant, PhD, Janani Parameswaran, PhD, and Jie Jiang, PhD in the Department of Cell Biology at Emory University School of Medicine, through a close collaboration with Lu Rao, PhD, and Arne Gennerich, PhD, at Albert Einstein College of Medicine, and Rüstem Yilmaz, PhD, and Jochen Weishaupt, MD, at Heidelberg University.
Other collaborators involved in this study include Gary Bassell, PhD, and Jonathan Glass, MD, from Emory University, and Isabel Loss and Philipp Koch, PhD, from Heidelberg University.
Next steps in this research include looking at downstream molecular mechanisms underlying toxicity from the KIF5A mutant protein that causes ALS. Currently, there is no cure for ALS, nor any effective treatment to slow down its progression. Study authors say their research identifying the presence of KIF5A ALS variants will facilitate the development of new therapeutic approaches targeting axonal transport using different model systems.
The research was supported by the ALS Association, DFG, the National Institute of Aging and the National Institute of Neurological Disorders and Stroke.