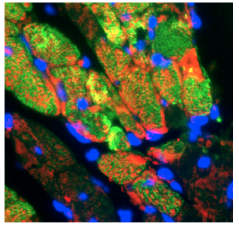
Forty-eight hours after an artificial heart attack in mice, the growth factor IGF-1 (red) can be seen in the heart muscle in response to the injury.
The enzyme MMCP-4 or chymase appears to limit the beneficial effects of IGF-1.
Cardiologists generally agree that if someone has a heart attack, the window of opportunity to intervene closes within hours, because heart muscle dies and doesn’t come back. A longer period may exist when it is feasible to amplify the heart’s attempts to heal itself, researchers from Emory University School of Medicine have discovered.
Hints about this possibility came from studying mice lacking the enzyme MMCP-4 (mouse mast cell protease 4 or chymase), which experience less damage to their hearts after an artificial heart attack. The differences between these mice and regular mice are not visible a day after the heart attack, but are substantial several days later.
The results were published Monday, June 6 in Proceedings of the National Academy of Sciences.
"In our experiments, if you look at the heart 24 hours after blood flow is restored, there’s a large area at risk.," says senior author Ahsan Husain, PhD, professor of medicine (cardiology) at Emory University School of Medicine. "Many of the heart muscle cells are not dead yet, but without intervention many of these cells will die over the next few days. We have found a way of saving them from this late death by improving their environment."
"It is widely believed that heart muscle cells die between minutes and hours after the blood supply is restored," says Nawazish Naqvi, PhD, assistant professor of medicine (cardiology) at Emory University School of Medicine, whose laboratory partnered with Husain’s. "Our findings show that a substantial level of heart muscle cell death occurs few days later, and its prevention by suppressing chymase activity could dramatically improve outcomes."
MMCP 4 appears to limit the heart’s attempts to heal itself. It chews up a growth factor called IGF-1 produced after the heart is damaged, Husain, Naqvi, and their colleagues showed. The first authors of the paper are cardiology fellow Thor Tejada, MD and postdoctoral fellow Lin Tan, PhD.
MMCP-4, known as chymase in humans, was already known to act as an "angiotensin converting enzyme." An important class of blood pressure medications (ACE inhibitors) block activity by a similar enzyme that generates the blood pressure regulator angiotensin II.
After a heart attack, patients are often prescribed ACE inhibitors or ARBs (angiotensin receptor blockers) because they reduce stress on the heart. Several pharmaceutical companies were developing chymase inhibitors but lost interest a decade ago when clinical studies suggested that blocking chymase wouldn’t provide any additional benefits through blood pressure control.
"I had wanted to nail down this loose end," Husain says. "And we found that chymase is not important for its angiotensin II effects. But that doesn’t mean chymase is not important."
The findings suggest that chymase inhibitors could achieve effects similar to those seen in mice lacking MMCP-4. Two weeks after artificial heart attack, those mice had left ventricular ejection fractions (a measure of the heart’s pumping power) that were 26 percent higher than controls.
Chymase inhibitor drugs would work by allowing IGF-1, produced by the injured heart, to last longer. In mice, IGF-1 levels normally peak at around 48 to 72 hours after heart damage, and the Emory researchers showed that MMCP-4 is the major enzyme that degrades it.
A clinical trial using a chymase inhibitor in heart failure patients is underway in Europe. Husain and Naqvi say that, based on their findings, the optimal trial should be in patients soon after a heart attack. IGF-1, exogenously supplied, has itself been tested in a clinical trial and shown to spur heart muscle growth. Husain and Naqvi say an indirect, drug-mediated approach may have advantages of increasing local, cardiac IGF concentrations in a more sustained way.
This work was funded by grants from the National Heart Lung and Blood Institute (HL079040, HL127726, HL098481, T32HL007745, HL092141, HL093579, HL094373 and HL113452), the American Heart Association (13SDG16460006), the Swedish Research Council, and the National Health and Medical Research Council of Australia (573732). Additional support came from the Emory University Department of Medicine, Carlyle Fraser Heart Center and Emory University Hospital Midtown.