Cystic Fibrosis
WARRIORS
Aggressive defenses against this genetic disease are extending lifespans


Andy Lipman first researched cystic fibrosis (CF) when he was 9 years old, wanting to find out more about the disease he’d been diagnosed with at birth. He discovered that people with CF, a progressive, genetic disease that causes thickened mucus to form in the lungs and other organs, rarely lived to age 25.


Directly above: Andy Lipman (on his mother’s lap) was diagnosed with CF when he was born. His older sister died of CF as an infant. Top photo: Lipman, now 46, uses a percussive vest daily to ease his breathing. Photo by Jack Kearse.
Directly above: Andy Lipman (on his mother’s lap) was diagnosed with CF when he was born. His older sister died of CF as an infant. Top photo: Lipman, now 46, uses a percussive vest daily to ease his breathing. Photo by Jack Kearse.
“I was shocked,” he says. “My parents were always telling me, you’ll be fine. But here was the encyclopedia, telling me something else. Which was true? I got really upset.”
Lipman’s sister had died of CF before he was born, living only 16 days. “Growing up, I didn’t talk about my CF or let anyone in,” he says. “My parents told my teachers. I was embarrassed and afraid—I didn’t know how long I was going to live.”
His parents encouraged him to live a normal life and play sports, which “really helped me physically.”
He went to a pediatric CF clinic for his care all the way through college. “There were no adult clinics back then,” he says. “Adult CF was an oxymoron. People just didn’t live that long.”
Lipman, now 46, has benefitted from advances in treatment and medications that have extended the median life expectancy of people with CF to 42 to 47 years, with some patients living far beyond that.
His doctor, Emory pulmonologist Randy Hunt, says CF has become a chronic—if still fatal—illness.
“It’s still a tough disease to be born with,” Hunt says. “When you’re very young, your parents can make you do all the right things. As you get older and go to school, you have to take handfuls of pills every time you eat, wear a mask at times, and use a vest for high-frequency chest wall oscillation…things that detract from your social experiences. Early adulthood can be a really difficult time. Deaths for CF patients still peak in their 20s and 30s.”
Indeed, in college, Lipman experienced depression and anxiety.
He had to carry his medications everywhere. The percussion vest he used each day to break up the mucus in his lungs was big and loud. “It was hard to feel normal, like I fit in,” he says. “I stopped doing treatments or taking my medications. I was skinny and felt ugly.” He stopped going to classes, and seldom left his room on weekends.
“My lung function went down pretty severely. Things were falling apart,” he says. “But then I got mad at myself and turned it around.” Lipman took a weightlifting class, got stronger, and went from 100 pounds to 165 pounds. He did his treatments every day.
“Living life requires more than just breathing. It requires loving every minute we have on this earth,” Lipman says.


New medications and therapies are helping CF patients lead longer, healthier lives—a feat that still requires a lot of care and self-discipline.
There is still no cure for CF, but there are drugs to help thin and clear the thick mucus from the airways, enzymes to help digest food, and antibiotics to prevent and treat infections. Recently, medications tuned to the genetic mutations that cause the disease have helped people with CF experience fewer flare-ups and hospitalizations and improved lung function.
Inhaling hypertonic saline as well as other medications through a nebulizer helps with breathing problems. “Most patients use the nebulizer two times a day for at least an hour, and sometimes up to four times,” Hunt says. “People think of CF as a lung disease, but really it is a systemic illness, impacting a patient’s bones, reproductive system, GI tract, pancreas, and other internal organs. Many patients need to eat 3,000 calories a day just to maintain their weight, and they have to take one to 10 pills every time they eat a meal,” says Hunt, an assistant professor of medicine at Emory who completed his fellowship in pulmonary and critical care medicine at Emory in 2013, and stayed to practice.
Hunt says they see about 318 patients in Emory’s Adult Cystic Fibrosis program. For outpatient care, patients are seen at the Emory Clinic. Pulmonary function testing and visits with each member of the CF care team take place within the patient’s exam room.
“We are set up as their health care home,” says Hunt, medical director of the Adult CF clinic. “For instance, patients may have endocrine problems, and we have a great endocrinologist. They also have higher rates of anxiety and depression, so we offer mental health counseling.” Also on staff are a GI specialist, a dietician, three nurses, a social worker, and two respiratory therapists.
There is no waiting room, since patients with CF cannot be closer than six feet from each other because they can carry bacteria in their lungs that could be harmful to someone else with CF, as reflected in the recent movie/teen love story Five Feet Apart. There is also a complex protocol to disinfect the exam rooms, Hunt says. “Our patients don’t present a danger to people without CF, only to each other,” he says. “It’s a socially isolating disease—you can’t be around anyone else who understands exactly what you’re going through. Social media has been wonderful for people with CF.”


“Living life requires more than just breathing. It requires loving every minute we have on this earth.”
–Andy Lipman


Because of the seriousness of the disease, the clinic sees CF patients every three months even when they are doing well. Patients often start out in the Children’s and Emory Pediatric Program and transition to the Emory Adult Program by the time they turn 21. The Children’s and Emory CF Care Center, which encompasses both programs, is one of the largest in the country. The Care Center follows about 375 pediatric CF patients, says director Rachel Linnemann, an Emory assistant professor of pediatrics. “Starting as young teens, we teach them the skills and knowledge they need to succeed in adult care,” says Linnemann.

Randy Hunt, head of Emory’s Adult CF program, says his patients are living longer and better, but “it’s still a tough disease to be born with.” Photo by Jack Kearse.
“For instance,” says transition coordinator and nurse practitioner Brandi Middour-Oxler, “we have teens take a nutrition quiz to ensure they understand why they take certain medications and supplements with meals.”
“This is a high-risk time for our patients, we call it the ‘adolescent slide,’ ” says Linnemann. “We want to help them understand the rationale behind their treatments, the why.”
About 85 percent of the CF center’s patients participate in research, whether donating blood or mucus samples or participating in clinical trials. “We’re an active site for trials, and have tested several classes of the new modulators, including the combination of lumacaftor and ivacaftor (Orkambi), tezacaftor and ivacaftor (Symdeko), and the new triple combination elexacaftor/tezacaftor/ivacaftor (Trikafta),” Linnemann says. “We’re seeing these drugs work for many patients, with improvements in lung function and less hospitalizations. Eventually 90 percent of people with CF could benefit from modulator therapy.”
Pediatric deaths from CF have gone down considerably in the past decade. “It is fairly rare to die younger than 21 in our program,” she says. “More than half of people living with CF are now adults.” Indeed, the median age of death was 30.8 years for those who died from complications of CF in 2018. Even more hopeful, the median predicted survival of an infant born with CF in 2018 is 47.4.
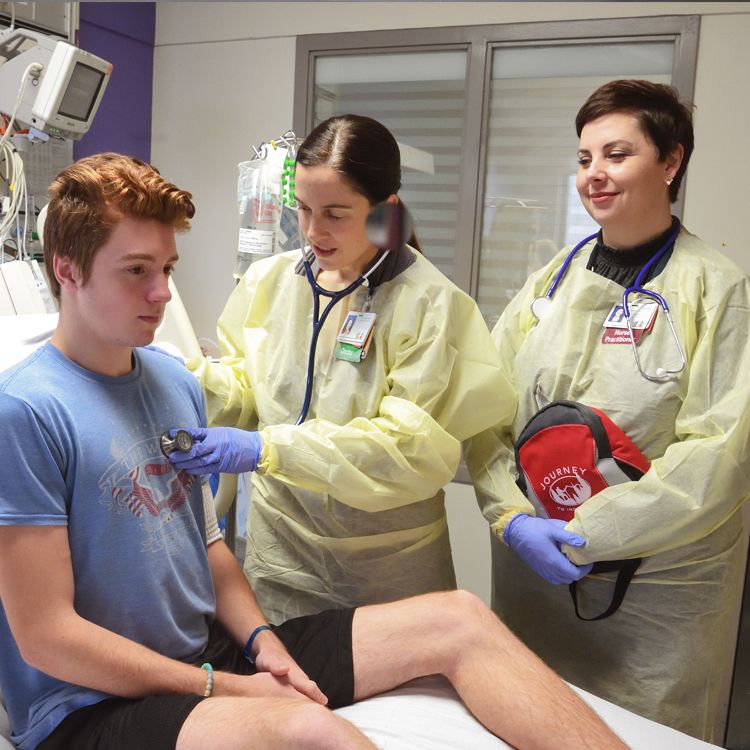
Corbin Heyl, 19, is beginning preparations to move to Emory’s Adult CF Program with help from Rachel Linnemann, head of the Pediatric CF Program, and transition coordinator Brandi Middour-Oxler. Photo by Jack Kearse.
The increasing life expectancy is good news for Corbin Heyl, 19, a Children’s and Emory CF patient whose younger brother Luke, 17, also has CF. Corbin, who is at Children’s Healthcare of Atlanta for a check-in because his lung function dropped and traces of pneumonia were detected, says, “CF doesn’t really scare me. It’s just inconvenient.”
The brothers are planning to transition from the pediatric CF program to the Emory adult program together, and are hopeful about new medications.
Corbin says he and Luke are pretty good about staying the required distance apart (“It’s just kind of a habit in our house”) unless they are playing basketball. “When we guard each other, we get pretty close,” he admits.

Andy Lipman, 46, who has CF, uses a percussive vest daily to ease his breathing. He has outlived every prediction due to new drugs and therapies. Photo by Alyssa Pointer, AJC via AP.
Sports and physical fitness are a way for CF patients to be proactive about their health. Lipman, now a husband and father of two living in Sandy Springs, is a Little League coach and avid runner. “I run a half hour a day and do the Peachtree every year,” Lipman says. “And I see a personal trainer a couple of times a week.” To honor the brief life of his sister, he hosts a popular “Wish for Wendy” softball tournament in Alpharetta each fall, and has raised more than $4.5 million over 20 years for the CF Foundation. “So many new drugs are out, with more clinical trials and breakthroughs coming,” he says. “Treatments take less time and are lighter and more portable. It’s easier to travel. You can definitely live a more normal life.”
Growing up, Lipman says, he had no CF role models or sources of inspiration, but that has changed. He has completed his fourth book, The CF Warrior Project: 65 Stories of Triumph against Cystic Fibrosis (cfwarriorproject.org). “All around the world,” he says, “people who have CF who are doing amazing things: a circus acrobat, a mountain climber, a firefighter. They’re all ages, from teens to 70s.”
And they are fathers. Although Lipman, like the vast majority of men with CF, lacks a vas deferens, he and his wife (who is not a carrier) were able to have children through in vitro fertilization. “We have a terminal, invisible disease,” he says. “We need hope raising, to believe life is there for the taking, regardless of the hand you are dealt.”
Story by Mary Loftus

Randy Hunt, head of Emory’s Adult CF program, says his patients are living longer and better, but “it’s still a tough disease to be born with.” Photo by Jack Kearse.
Randy Hunt, head of Emory’s Adult CF program, says his patients are living longer and better, but “it’s still a tough disease to be born with.” Photo by Jack Kearse.

Corbin Heyl, 19, is beginning preparations to move to Emory’s Adult CF Program with help from Rachel Linnemann, head of the Pediatric CF Program, and transition coordinator Brandi Middour-Oxler. Photo by Jack Kearse.
Corbin Heyl, 19, is beginning preparations to move to Emory’s Adult CF Program with help from Rachel Linnemann, head of the Pediatric CF Program, and transition coordinator Brandi Middour-Oxler. Photo by Jack Kearse.

Andy Lipman, 46, who has CF, uses a percussive vest daily to ease his breathing. He has outlived every prediction due to new drugs and therapies. Photo by Alyssa Pointer, AJC via AP.
Andy Lipman, 46, who has CF, uses a percussive vest daily to ease his breathing. He has outlived every prediction due to new drugs and therapies. Photo by Alyssa Pointer, AJC via AP.
